
| EU GMP Part IV – ATMPs GMP要点解析(二) |
| 作者:admin 来源:原创 发布日期:2022/4/18 17:35:34 点击次数:3548 |
|
|
EU GMP Part IV – ATMPs GMP要点解析(二)
转载内容。 非原创和整理
补充 a) 欧盟2018年6月发布的Annex 2 Manufacture of Biological active substances and Medicinal Products for Human Use不再包含有关ATMPs的内容。 b) 通过对ATMPs GMP的研读,这份GMP的无菌管理部分参考或引用了很多欧盟在2017年12月发布的Annex 1 Manufacture of Sterile Medicinal Products征求意见稿中的内容。整体要求高于现行版Annex 1。 2. Risk-based approach 这一章与EU GMP不同,EU GMP将风险管理的内容放在了“第一章 药品质量体系”中,详细内容则描述为参见ICH Q9。在ATMPs GMP中使用了很大的篇幅结合此类产品的特点对风险管理的内容进行了详细的描述。 2.1. Introduction 2.10.ATMPs are complex products and risks may differ according to the type of product, nature/characteristics of the starting materials and level of complexity of the manufacturing process. It is also acknowledged that the finished product may entail some degree of variability due to the use of biological materials and/or complex manipulation steps (e.g. cultivation of cells, manipulations that alter the function of the cells, etc.). In addition, the manufacture and testing of autologous ATMPs (and allogeneic products in a donor-matched scenario) poses specific challenges and the strategies implemented to ensure a high level of quality must be tailored to the constraints of the manufacturing process, limited batch sizes and the inherent variability of the starting material. 简译: 1、根据产品类型、起始物料的性质/特征以及生产工艺的复杂水平,ATMPs的风险也有所不同。 2、由于生物材料的应用和/或复杂的操作步骤(例如细胞培养,改变细胞功能的操作等等),成品也许会具有某种程度的可变性。 3、此外,自体ATMPs(和供体匹配情形下的同种异体产品)的生产和检验具备特有的难点,所实施的用以保证高质量水平的策略必须根据生产工艺的限制、有限的批量以及起始物料的变异性来制定。 总结观点: 1、这一条介绍了ATMPs的特点,由于生物技术产品的不确定因素与化学药品相比更多,因此控制策略需要根据法规要求以及风险评估的结果来确定。 2、生物制品方面使用风险管理工具最直接的例子就是在中国药典三部中对原材料、辅料通过风险管理进行分级。 2.11. ATMPs are at the forefront of scientific innovation and the field is experiencing rapid technological change that also impacts on the manufacturing processes. For instance, new manufacturing models are emerging to address the specific challenges of ATMPs (e.g. decentralised manufacturing for autologous products). Additionally, ATMPs are also often developed in an academic or hospital setting operating under quality systems different to those typically required for the manufacture of conventional medicinal products. 简译: 1、ATMPs位于科技创新的前沿,这个领域快速的技术变革也影响了生产工艺。例如,新生产模式不断涌现,以应对ATMPs的特殊挑战。(例如自体产品的分散化生产) 2、此外,ATMPs也经常在学院或医院环境下进行开发,其运行遵照的质量体系也不同于传统药品生产的通行要求。 总结观点: 1、从生产技术上来讲,目前的细胞培养以及后续的工艺技术都有非常快速的变化,例如从可反复使用的生物反应器到一次性用品的大量普及应用,可以说生物制品在技术革新方面已经位于顶端。 2、与传统产品不同,细胞治疗在以前是作为一项治疗技术来应用,因此其开发环境往往是在医院或学校。而医院的质量体系与药品的质量体系有非常大的区别,医院的质量管理关注点是围绕着医疗相关的各个要素,包括诊断、治疗、护理、卫生、药剂等方面。该GMP引言部分提出了更为严格的要求,未来在医院进行的此类产品的生产也要与上市产品的生产条件等同或相同,而不是只具备医疗条件。 2.12. It follows that, in laying down the GMP requirements applicable to ATMPs, it is necessary to recognise a certain level of flexibility so that the ATMP manufacturer can implement the measures that are most appropriate having regard to specific characteristics of the manufacturing process and of the product. This is particularly important in the case of investigational ATMPs, especially in early phases of clinical trials (phase I and phase I/II), due to the often incomplete knowledge about the product (e.g. potency) as well as the evolving nature of the routines (in order to adjust the manufacturing process to the increased knowledge of the product). 简译: 1、在制定适用于ATMPs的GMP要求时,需要某种程度的灵活性以便于ATMP生产企业能够实施最适用于产品以及生产工艺特殊性的措施。 2、由于对产品(例如效价)的理解以及常规的演变特点(为了调整工艺以增加产品知识)还不够充分,这对临床用ATMPs尤其重要,特别是临床试验的早期阶段(一期或一期/二期)。 总结观点: 这条阐释了制定ATMPs GMP的一些考虑。 2.2. Application of the risk-based approach by ATMP manufacturers 2.13. The risk-based approach (“RBA”) is applicable to all type of ATMPS. It applies in an equal fashion to all type of settings. The quality, safety and efficacy attributes of the ATMPs and compliance with GMP should be ensured for all ATMPs, regardless of whether they are developed in a hospital, academic or industrial setting. 简译: 1、RBA适用于所有类别的ATMPs。 2、所有ATMPs均应确保ATMPs的质量、安全性、药效等属性以及GMP符合,无论其在医院、学院或是工业环境中进行开发。
总结观点: 1、指出了基于风险方法的适用范围。 2、强调了无论在何种环境开发,最终要达到的目标是一致的,即确保实现药品的固有属性以及GMP的符合性。这里实际是QbD理念的问题,也就是说药品的属性和GMP符合性要在产品设计阶段就要加以考虑和计划,而不能全部都放到商业化阶段才去考虑。例如很多前沿治疗药物以前或当前的最终运输包装采取的是微量离心管,如下图。这种类型的离心管一般只用于离心之用而不作为产品的最终包装。对于用作产品的内包装材料而言,需要考虑化学相容性、产品相容性、析出物等等因素,还需要考虑无菌包装密封性。这些都要在设计阶段进行考虑或试验。
2.14. Manufacturers are responsible for the quality of the ATMPs they produce. The risk-based approach permits the manufacturer to design the organisational, technical and structural measures that are put in place to comply with GMP -and thus to ensure quality according to the specific risks of the product and the manufacturing process. While the risk-based approach brings flexibility, it also implies that the manufacturer is responsible to put in place the control/mitigation measures that are necessary to address the specific risks of the product and of the manufacturing process. 简译: 1、生产企业要对所生产的ATMPs质量负责。 2、RBA允许生产企业按照产品和工艺的特殊风险设计、实施组织化、技术化和结构化的措施,以符合GMP并确保质量。 3、尽管RBA提供了灵活性,但也表明生产企业有责任实施必要的控制/缓解措施以处理产品和工艺的特殊风险。
总结观点: 1、强调了生产企业的责任。无论在医院生产还是在商业化规模的工厂生产,都要负责产品质量。 2、为了符合GMP和保证产品质量,允许生产企业基于风险评估采取针对性的措施。例如: 基于CQAs和CPPs的评估建立控制策略; 根据对原辅料的风险评估采取不同的检验和审计策略; 基于产品工艺特点和对其进行的风险评估,考虑采取隔离器(Isolator)还是生物安全柜进行操作。(如果法规明确规定操作背景及环境则不能通过风险评估降低要求) 2.15. The quality risks associated with an ATMP are highly dependent on the biological characteristics and origin of the cells/tissues, the biological characteristics of the vectors (e.g. replication competence or reverse transcription) and transgenes, the level and characteristics of the expressed protein (for gene therapy products), the properties of other non-cellular components (raw materials, matrixes), and the manufacturing process. 简译: 与ATMP相关的质量风险高度依赖于: 1、细胞/组织的生物学特性和来源; 2、载体的生物学特性(例如复制能力和逆转录); 3、转基因; 4、表达蛋白的水平和特性(基因治疗产品); 5、其他非细胞成分的性质(原材料,基质); 6、生产工艺。
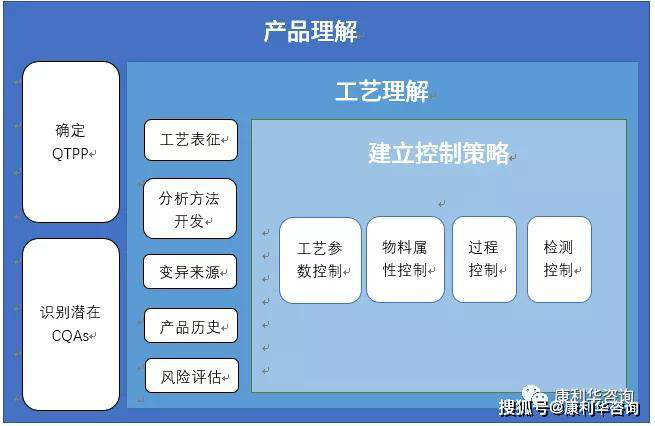
总结观点:阐述了质量风险管理要依据的要素。 2.16. When identifying the control/mitigation measures that are most appropriate in each case, the ATMP manufacturer should consider all the potential risks related to the product or the manufacturing process on the basis of all information available, including an assessment of the potential implications for the quality, safety and efficacy profile of the product, as well as other related risks to human health or to the environment. When new information emerges which may affect the risks, an assessment should be made whether the control strategy (i.e. the totality of the control and mitigation measures applied) continues to be adequate. 简译: 1、在识别每种情形下更适用的控制/缓解措施时,ATMP生产企业应当基于所有可获取的信息考虑所有与产品或生产工艺相关的潜在风险,包括对产品质量、安全性和疗效概况潜在影响的评价,以及对人类健康或环境的其他相关风险。 2、当新信息可能影响风险时,应当评价控制策略(即所采取的全部控制和缓解措施)是否仍然足够。 总结观点:描述了风险评估的基础以及风险评估的范围。 1、基础:所谓“可获取的信息”,是指可支持风险评估过程的各种数据与信息文献等。例如要对工艺参数进行关键性评估,应当具有工艺规程、工艺参数列表、研发报告与数据、有关的变更或偏差数据等,而非纯粹的“头脑风暴”; 2、范围,产品及工艺相关 对产品质量、安全性和疗效概况潜在影响的评价; 对人类健康或环境的其他相关风险; 3、持续性评估以及策略更新:随着企业对生产工艺和质量的知识的积累,对工艺和质量的理解不断加深,会产生或获得新的知识与信息,这个时候需要评价是否会对已形成的控制策略造成影响或已有控制策略是否足以应对产生的新情况。例如在研发阶段评估以及基于当时研发的技术手段,某个工艺参数被判定为“关键”,随着科技发展、工艺知识积累与更新以及新技术的应用(比如PAT的应用,一次性技术的实施),这个参数在重新评价时也许会被定性为“非关键”,此时要考虑基于以前评价得到的策略是否依然适用,反之亦然。 2.17. The evaluation of the risks and the effectiveness of the control/mitigation measures should be based on current scientific knowledge and the accumulated experience. Ultimately, this evaluation is linked to the protection of patients. 简译: 风险评价以及控制/缓解措施的有效性应当基于现有的科学知识以及积累的经验。最重要的是,评价要与对患者的保护联系起来。 总结观点:仍然强调了基础。 1、风险评价和措施的有效性评价应当是基于事实和数据的,不是“空想主义”。通过风险评价得到的风险因素,对于高或中度风险水平的因素要采取措施降低风险水平,这个措施的有效性并不是只在风险评估的记录上想当然的标注为“低”就结束了,要通过实施后的结果来判定这个或这些措施是否有效,也就是说还要注意不要丢掉了“风险回顾”这个过程。 2、以患者安全为中心是最重要的基础。 2.18. The level of effort and documentation should be commensurate with the level of risk. It is neither always appropriate nor always necessary to use a formal risk management process (using recognized tools and/ or internal procedures e.g., standard operating procedures). The use of informal risk management processes (using empirical tools and/or internal procedures) can also be considered acceptable. 简译: 1、投入与文件水平应当与风险级别相当。 2、运用正式的风险管理过程(使用公认的工具和/或内部程序,例如SOP)并不总是恰当和必要的。非正式的风险管理过程(使用经验工具和/或内部程序)也是可以得到认可的。 总结观点: 风险管理的运用是国内很多生产企业的难点。例如以下是经常出现的问题: 1、不考虑评估对象的复杂性,只要涉及风险评估,一律使用FMEA作为工具; 2、进行风险分级的时候,全部采取打分的方式进行; 3、为了不做某件事,通过风险评估强行评估为低风险; 4、风险管理流程不完整,只有一张FMEA的大表,没有风险识别过程,没有风险回顾和风险沟通过程。 2.19. The application of a risk-based approach can facilitate compliance but does not obviate the manufacturer's obligation to comply with relevant regulatory requirements and to demonstrate that it is able to adequately manage the risks of the product/manufacturing process. It likewise does not replace appropriate communications with the authorities. 简译: RBA的运用可以有利于法规符合性,但并不免除生产企业符合有关法规要求以及证明其有能力充分地管理产品/生产工艺的风险的义务。同样也不能取代与官方进行适当的沟通。 总结观点: 1、如果法规上有明确的条款要求,那么按照法规执行,不需要风险评估。例如非最终灭菌产品的无菌灌装环境在条款里明确规定为B级背景下的A级,我们就不需要评估背景不是A级会造成什么样的风险; 2、如果法规上没有明确的要求,而在非官方组织的某个指南中有描述,可以采用,但采用之前要进行风险评估。 3、以上是基于法规和指南的法律顺位而得到的结论,国内的法律效力排序由高到低依次是宪法、基本法、一般性法律、行政法规、条例,非官方组织或机构发布的指南是没有法律效力的,只能作为某个或某些主题的参考。 Investigational ATMPs 2.20. The application of GMP to investigational ATMPs is intended to protect the clinical trial subjects and it is also important for the reliability of the results of the clinical trial, in particular by ensuring consistency of the product, that the results of the clinical trial are not affected by unsatisfactory manufacturing used and that changes of the product throughout the development are adequately documented. 简译: 临床用ATMPs实施GMP是为了保护临床试验受试者,对临床试验结果的可靠性也很重要,特别是通过确保产品的一致性,临床试验的结果不会因不符合要求的生产而受到影响,并恰当的记录了开发过程中发生的产品变更。 总结观点:强调了在临床用药生产过程中实施GMP的目的和重要性。 1、目前在药品研究、商业化生产、退市等各个阶段的管理主要是以下几个方面: 临床试验:GCP; 非临床研究:GLP; 药品的商业化生产:GMP; 药品流通:GDP/GSP; 临床用药的生产与质量管理:符合GMP条件;(FDA有专门针对一期临床样品生产的CGMP) 中试且用于注册申报:符合GMP条件; 研发实验室:目前没有具体使用某个规范的要求,总体要求是要建立质量体系; 2、符合GMP条件:即按照GMP的要求进行管理,不强制通过GMP认证。 2.21. It is important to ensure that data obtained from the early phases of a clinical trial can be used in subsequent phases of development. Therefore, a functional quality system should be in place for the manufacturing of investigational ATMPs. 简译: 重要的是确保临床试验早期阶段获得的数据能用于开发的后续阶段。因此,临床用ATMPs应当运行质量体系。 总结观点: 1、强调了临床用ATMPs的生产与质量管理应当建立质量体系; 2、临床试验获得的数据会用于指导研发,例如局部的工艺参数调整。这时候要确保数据的可用性或者说要符合数据完整性/可靠性的所谓ALCOA+原则。 2.22. The quality and safety of the product needs to be ensured from the first stages of development. Nevertheless, it is acknowledged that there is a gradual increase in the knowledge of the product and that the level of effort in the design and implementation of the strategy to ensure quality will step up gradually. It follows that the manufacturing procedures and control methods are expected to become more detailed and refined during the more advanced phases of the clinical trial. 简译: 产品的质量和安全性需要在开发的最初阶段就得到保证。然而事实上产品知识是逐步递增的,在质量保证策略的设计以及实施中投入的水平将会逐步增加。因此在临床试验的进展期中,生产程序以及控制方法可能会更为细化和完善。 总结观点: 1、实质上是强调了QbD的理念。 2、随着研究的逐步深入,产品知识和工艺理解逐步累积起来,质量保证策略和控制策略会越来越完善,相应的生产工艺、IPC、检验方法也会最终确定并形成具有指导意义和可操作性的各类文件。 3、控制策略的建立简图(译自PDA TR81)
2.23. While the responsibility for the application of the risk-based approach lies with the manufacturer, it is encouraged that the advice of the competent authorities is sought in connection with the implementation of the risk-based approach for investigational ATMPs and, in particular, regarding early phases of clinical trials. The application of the risk-based approach should be consistent with the terms of the clinical trial authorisation. The description of the manufacturing process and process controls in the clinical trial authorisation application should explain, as appropriate, the quality strategy of the manufacturer when the risk-based approach is applied. 简译: 1、尽管使用RBA是生产企业的责任,但鼓励在临床用ATMPs实施RBA方面寻求官方的建议,特别是在临床试验的早期阶段。 2、RBA的使用应与临床试验许可的条款一致。必要时,临床试验许可申请中的生产工艺以及过程控制的描述应解释使用RBA时采取的质量策略。 总结观点: 1、每个组织、每个部门、每个人对法规的理解是不一样的,对于哪些步骤、哪些方面可以运用RBA的方式,除了自行按照法规要求来实施之外,还要征求官方的建议,尤其是一些法规中没有详细描述的地方或是企业理解的不透彻、不清楚的地方,以避免将来的审评出现不可弥补的缺陷。 2、强调了使用RBA时不能违背临床试验许可的要求。同时要求,在申请临床试验许可时递交的资料中应当对使用RBA所采取的质量控制和保证策略进行阐述,换言之,不应只递交工艺步骤、工艺参数和IPC,应当将RBA输出的控制措施一并递交。 2.24. For aspects that are not specifically covered by the clinical trial authorisation, it is incumbent upon the manufacturer to document the reasons for the approach implemented and to justify that the totality of the measures applied are adequate to ensure the quality of the product. In this regard, it is recalled that alternative approaches to the requirements explained in these Guidelines are only acceptable if they are capable of meeting the same objective. 简译: 1、对临床试验许可没有明确涵盖的各个方面,生产企业有义务用书面证明方法实施的原因并论证采取的全部措施足以保证产品质量。 2、从这个角度来讲,指南中解释的需求的其他方法仅在能够符合相同目标的情况下才可使用。 总结观点: 临床试验许可的批准中会列明一些要求,但不可能将所有的要求全部列举进去。因此如果企业使用了RBA的方式建立了策略并采取了对应的措施,应当有书面的证明资料,包括验证/确认等。 Authorised ATMPs 2.25. For authorised ATMPs, the application of the risk-based approach should be consistent with the terms of the marketing authorisation. When providing the description of the manufacturing process and process controls in the marketing authorisation application (or, as appropriate, in the context of the submission of a variation), account can be taken of the specific characteristics of the product/manufacturing process to justify adaptation/deviation from standard expectations. Thus, the strategy to address specific limitations that may exist in connection with the manufacturing process, including controls of raw materials and starting materials, the manufacturing facilities and equipment, tests and acceptance criteria, process validation, release specifications, or stability data should be agreed as part of the marketing authorisation. 简译: 1、对authorised ATMPs而言,RBA的运用应当与上市许可的条款一致。 2、上市许可申请中(或者,递交的变更文件)提供生产工艺和过程控制的描述时,要考虑产品/生产工艺的特殊性质以论证与标准预期的适配/偏离。因此用于处理可能与生产工艺有关的特殊限制的策略应获准并作为上市许可的组成部分,包括原材料与起始物料的控制,生产设施和设备,检验和可接受标准,工艺验证,放行标准或稳定性数据。 总结观点: 1、对上市许可ATMPs使用RBA的原则性要求。 2、与前文述及内容的原则类似,要论证RBA的使用以及因之得到的策略与措施。 2.26. For aspects that are not specifically covered by the marketing authorisation, it is incumbent upon the manufacturer to document the reasons for the approach implemented when the risk-based approach is applied, and to justify that the totality of the measures applied are adequate to ensure the quality of the product. In this regard, it is recalled that alternative approaches to the requirements explained in these Guidelines are only acceptable if they are capable of meeting the same objective. 简译: 上市许可中没有明确涵盖的各个方面,当使用RBA时,生产企业有义务用书面证明实施RBA的原因并论证采取的全部措施足以保证产品质量。 从这个角度来讲,指南中解释的需求的其他方法仅在能够符合相同目标的情况下才可使用。 2.3. Examples of the application of the risk-based approach 2.27. This Section contains a non-exhaustive list of examples to illustrate some of the possibilities and limitations of the risk-based approach. 简译: 本节包含用于解释RBA的一些可能性与限制的示例清单。 2.3.1. RBA in connection with raw materials 原材料相关的RBA 2.28. The application of the risk-based approach when determining the strategy to ensure the quality of the raw materials is explained in Section 7.2. 简译: 确定原材料质量保证策略时对RBA的运用参见7.2. 2.29. The application of the risk-based approach requires that the manufacturer has a good understanding of the role of the raw material in the manufacturing process and, in particular, of the properties of the raw materials that are key to the manufacturing process and final quality of the product. 简译: 使用RBA需要生产企业对原材料在生产工艺中的作用有良好的认识,特别是对生产工艺和产品的最终质量有重要影响的原材料特性。 总结观点: 1、使用RBA的前提条件或者基础。 2、质量风险管理应当与产品的关键质量属性相关联以保证不会对患者造成安全影响。对产品质量属性的评价可以至少得到CQAs(Critical Quality Attributes)与CMAs(Critical Material Attributes),前者是产品本身的CQA,后者是所使用原材料或活性成分的关键属性,例如活性成分的粒度、晶型、微生物质量等等。根据产品剂型的不同,所评价得到的原材料的CMA也会有所不同。 3、细胞培养常用的原材料包括培养基、缓冲液、添加成分(血清、因子、抗生素)等,企业需要根据研发经验、文献等结合运用风险评估的工具识别所使用原材料的哪些属性或特性会对细胞培养步骤造成重要影响。例如细胞培养步骤或工序依赖于溶液的渗透压、pH,提供的气体环境,无菌条件等因素,如果结合这些因素去评价培养基,那么培养基的无菌性、pH、适用性则是要重点关注的方面。 2.30. Additionally, it is important to take into account the level of risk of the raw material due to the intrinsic properties thereof (e.g. growth factors v. basic media, culture media containing cytokines v. basal media without cytokines, raw material from animal origin v. autologous plasma, etc.), or the use thereof in the manufacturing process (higher risk if the raw material comes into contact with the starting materials). 简译: 此外,重要的是要考虑内在特性(例如生长因子v.基础培养基,包含细胞因子的培养基v.不含细胞因子的基础培养基,动物源原材料v.自体血浆等等)或在生产工艺中的用途(若原材料与起始物料接触则为更高级别的风险)相关的原材料的风险级别。 总结观点: 1、这一条是对上一条(2.29)的进一步说明。 2、2.29说明了当使用RBA确定原材料的控制策略时要考虑原材料在工艺中的用途以及原材料特性是否会对工艺或产品质量造成重要影响,这一条进一步说明还要考虑原材料的风险级别。 来源:生物原材料、化学原材料; 批准情况:有上市许可、有国家标准、非药用; 安全:TSE/BSE,毒性化学物质、牛血清、水解酶; 用途:生产用培养基成分、细胞消化、蛋白质水解等; 3、与这一条最相近的就是中国药典三部中的“生物制品生产用原材料及辅料质量控制规程”,这篇规程中对原材料、辅料的分级质控做出了要求,根据原材料/辅料的来源、生产以及对生物制品潜在的毒性/安全性和外源因子污染风险等,按照风险等级由低到高分为四级。 2.31. Finally, it needs to be assessed if the control strategy (e.g. qualification of suppliers, performance of suitable functional testing, etc.) is sufficient to eliminate the risks or to mitigate them to an acceptable level. 简译: 需要评估控制策略(例如供应商的确认,适当的功能检查的性能等等)是否足以消除风险或使之降低到可接受的水平。 总结观点: 1、根据2.28~2.30的要求实施而得到的控制策略,例如关键项目(鉴别、微生物限度、细菌内毒素等)的检测、供应商审计、外源因子检测等,也需要进行风险评估。 2、实施评估的次序如下: a. 列明原材料和辅料清单; b. 根据原材料和辅料在工艺中的用途、对工艺和产品质量的影响,运用风险评估工具进行关键性评估与风险分级,得到每一种原材料/辅料的控制策略(级别与策略定义如中国药典三部所述); c. 对这些策略进一步评估,确定是否可接受。 2.3.2. RBA in connection with the testing strategy 检验策略相关的RBA 2.32. It is acknowledged that in some cases it may not be possible to perform the release tests on the active substance or the finished product, for example due to technical reasons (e.g. it may not be possible to perform the release tests on the combined components of certain combined products, time restrictions (i.e. the product needs to be administered immediately after completion of manufacturing), or when the amount of available product is limited to the clinical dose. 2.33. In these cases, an adequate control strategy should be designed. For example, consideration can be given to the following options: 简译: 1、某些情况下,可能无法执行对活性物质或成品的放行检验,例如由于技术原因(例如无法执行对组合产品的组份进行放行检验),时间限制(即产品需要在生产结束后立即给药),或可获得的产品量受限于临床剂量。 2、这种情况下,应当设计充分的控制策略。例如下列给出的考虑(2.34~2.36)。 总结观点: 1、组合产品:例如药械组合类型的产品(预灌装注射) 2、这两条是与细胞治疗、基因治疗等产品的使用特性有关联的。由于基于2.32举例的情况没有办法进行放行检验,无法确切的保证所生产出的产品一定能够符合既定质量标准,因此需要采取必要的控制措施来降低或消除风险,最大限度的保证患者的健康和用药安全。 2.34. • Testing of key intermediates (instead of the finished product) or in-process controls (instead of batch release testing) if the relevance of the results from these tests to the critical quality attributes of the finished product can be demonstrated. 简译: 重要中间产品的检验(非成品)或过程控制(非批放行检验):如果能够证明这些测试的结果与成品CQAs的相关性。 总结观点: 1、这一条有些类似中国GMP的第166条所述:“外购或外销的中间产品和待包装产品应当有质量标准;如果中间产品的检验结果用于成品的质量评价,则应当制定与成品质量标准相对应的中间产品质量标准。 2、例如,某个产品需要在制备后立即使用,这时产品无法进行效价测定。根据这一条,则可以对分装时或分装前的配制过滤工序进行取样测试。但要注意 a、取样对产品质量的影响,尤其是对无菌和细菌内毒素质量的影响; b、在这些步骤取样的样品能否顺利实施检验; c、如果测试项目与成品的CQAs相同,例如均为效价,那么检验方法应一致并应已经过验证; d、如果测试项目与成品CQAs不同,应有数据证明两者之间的关联性。 2.35. • Real time testing in case of short shelf-life materials/products. 简译: 实时检验:货架期短的物料/产品。 总结观点: 1、实时放行检验(Real time release testing,RTRT):The ability to evaluate and ensure the quality of in-process and/or final product based on process data, which typically include a valid combination of measured material attributes and process controls. (ICH Q8) 将被测量物料属性和工艺控制等的数据进行有效结合,据此评估和保证中间产品和/或最终成品质量的能力。 2、看似简单实际上实施起来还是有一定难度的,往往需要结合实时监测技术。实时放行检验需要结合对工艺的理解、控制策略并结合对有关产品质量的关键属性的at-line/on-line/in-line分析进行,要能够证明实时质量保证优于或者至少等同于以实验室为基础的样品检测。 术语对比:at-line/on-line/in-line/offline Guideline on process validation for finished products - information and data to be provided in regulatory submissions EMA/CHMP/CVMP/QWP/BWP/70278/2012-Rev1 27 February 2014 1、On-line: Measurement where the sample is diverted from the manufacturing process and not returned to the process stream。 检测样品从生产工艺中转移出来并不返回工艺流中。(例如水系统的在线TOC,TOC在线有旁路引出测定) 2、In-line: Measurement where the sample is analysed within the process stream and not removed from it. 样品在工艺流中进行检测并不从中移除。(例如CIP或水系统的在线电导率) 3、At-line: Measurement where the sample is removed, isolated from, and analysed in close proximity to the process stream. 样品从工艺流中移除、隔开,并且在靠近工艺流的地方分析的检测。(例如岗位监测pH) 4、Offline:离线(取样后在实验室测定样品的TOC) 2.36. • Increased reliance on process validation. When the scarcity of materials or the very short shelf-life limits the possibilities for release controls, the limitations should be compensated by a reinforced process validation (e.g. additional assays, such as potency testing or proliferation assays may be performed after batch release as supporting data for process validation). This may also be relevant for investigational ATMPs: while process validation is not expected for investigational medicinal products (see Section 10.3), it may be important when routine in-process or release testing is limited or not possible. 简译: 1、更多的依靠工艺验证。在样本量小或很短的货架期制约了放行控制的可能性时,应通过对工艺验证的扩充来弥补这种限制(例如额外的测试,例如在批放行后进行的效价测定或细胞增殖分析作为工艺验证的支持性数据)。 2、这也关系到临床用ATMPs:尽管未期望对临床用药进行工艺验证(参见10.3),但当日常过程检验或放行检验受到限制或无法进行时也许工艺验证就是重要因素。 总结观点: 1、这一条与常规执行的放行策略有所不同。常规执行的策略是:不能因工艺验证得到了符合要求的结果而取代放行测试。 2、这一条“先放行后测试”的做法实际上对患者而言是具有非常大的生命和健康风险的,但由于产品特点的制约,在充分进行评估之后可以实施。评估应当包括: a、根据具体制约放行控制的因素进行风险评估,确定是否有针对性的降低风险的措施; b、放行后进行的测试结果进行累积统计分析,用于支持工艺能力的数据。通过这些数据与工艺参数的关联,证明工艺能够保证产品质量的稳定与均一。 2.37. It is stressed that the release testing strategy should be performed in accordance with the marketing/clinical trial authorisation. 简译: 要强调的是,应当按照上市许可/临床试验许可执行放行检验策略。 总结观点: 这一条的言外之意是,如果要执行上述的策略应当在上市许可/临床试验许可时就申报并获得官方的批准。 2.38. The following examples may also be considered: 2.39~2.43为举例。 2.39. • The application of the sterility test to the finished product in accordance with the European Pharmacopoeia (Ph. Eur. 2.6.1) may not always be possible due to the scarcity of materials available, or it may not be possible to wait for the final result of the test before the product is released due to short shelf-life or medical need. In these cases, the strategy regarding sterility assurance has to be adapted. For example, the use of alternative methods for preliminary results, combined with sterility testing of media or intermediate product at subsequent (relevant) time points could be considered. 2.40. The use of validated alternative rapid microbiological methods may also be considered. For example, sole reliance on alternative microbiological methods according to Ph. Eur. 2.6.27 may be acceptable when this is justified having regard to the specific characteristics of the product and the related risks, and provided that the suitability of the method for the specific product has been demonstrated. 2.41. If the results of the sterility test of the product are not available at release, appropriate mitigation measures should be implemented, including informing the treating physician (see Section 11.3.2). 简译: 1、由于样本量小或由于短货架期/医疗需要在放行前不能等待检验的最终结果,不是总能够实现按照EP2.6.1进行成品的无菌检查。这种情况下,要采取无菌保证策略。例如,可以考虑对初步结果使用替代方法,与培养基或后续/相关时间点的中间产品的无菌检查相结合。 2、可考虑使用经验证的快速微生物方法。例如,如果产品的特性与相关风险得到了论证以及证明了方法适用性的情况下,按照EP2.6.27采用替代的微生物学方法也可以得到认可。 3、如果产品的无菌检查结果不能在放行时得到,应采用适当的降低风险的措施,包括通知治疗医师。 总结观点: 1、无菌检查,根据目前的各国药典,需要至少14天的培养时间。很多细胞类产品的有效期/货架期最短可能只有几个小时,因此在放行前无法等待无菌检查结果。但如果无菌检查不合格且已经用药,会对患者造成极大的健康安全风险,甚至危及生命。任何一个国家的GMP对无菌保证都给予了充分的重视,无论是何种类型的无菌产品。 2、由于取样的局限性,仅仅依靠无菌检查合格不是最可靠的无菌保证手段,在无菌制剂生产中,往往是基于无菌工艺设计、无菌操作、过程控制与检验结果相结合等措施来确保无菌产品的质量。 3、基于生物制品的特性,在欧盟GMP的Annex 2 以及本GMP(Part IV)描述了当无法在放行前得到无菌检查结果时的要求,例如对质量保证体系有效性的连续评估以及无菌保证策略,从体系保证以及过程控制的角度来确保无菌质量。根据需要包括但不限于以下的举例: a、对批生产记录的审核,包括环境监测结果等; b、产品检验结果和过程控制结果; c、快速微生物测定方法与中间产品无菌检查结果相结合; d、通知治疗医师。 2.42. • As cells in suspension are not clear solutions, it is acceptable to replace the particulate matter test by an appearance test (e.g. colour), provided that alternative measures are put in place, such as controls of particles from materials (e.g. filtration of raw material solutions) and equipment used during manufacturing, or the verification of the ability of the manufacturing process to produce low particle products with simulated samples (without cells). 简译: 由于细胞处于悬浮状态而不是澄清溶液,可以用性状检查(如颜色)替代颗粒物质检查。前提是采取替代措施,如控制源自物料(例如原材料溶液的过滤)和生产过程中所使用设备的颗粒或通过模拟样品(无细胞)证明生产工艺只产出少量颗粒产物的能力。 总结观点: 1、这一条与常规无菌制剂是不同的。常规无菌制剂的颗粒物质检查一般包括可见异物和不溶性微粒检查。 2、这一条基于细胞制品的特性提出了以性状检查替代颗粒物质检查,但是需要前提条件的,也就是说企业要根据产品以及评估、验证确定是否采用替代方法。从后半句来看,即便要使用替代方法也要先对物料、设备、工艺关于颗粒物质控制的能力进行验证。 2.43. • It may be justified to waive the on-going stability program for products with shorter shelf-life. 简译: 如有合理的理由,有效期很短的产品可免于持续稳定性程序。 总结观点: 1、根据ICH Q1的描述以及实践,稳定性考察主要有以下几种情况: a、新产品的稳定性研究; b、新产品上市后的持续稳定性考察; c、由于质量事件,例如偏差或变更导致的稳定性研究; 2、对于有效期/货架期短的产品,可能无法完全按照ICH Q1中要求的长期以及加速试验条件进行研究,例如时间和样品量的限制。 3、根据国内发布的细胞治疗制剂以及干细胞制剂的要求,需要根据产品的储存、使用以及运输情况考虑冻融试验(如果是冻存)、储存稳定性以及运输稳定性。 2.3.3. Additional considerations relevant for ATMPs that are not subject to substantial manipulation 2.44. Manufacturing processes of ATMPs not involving substantial manipulation of the cells/tissues are typically associated with lower risks than the manufacturing of ATMPs involving complex substantial manipulations. However, it cannot be inferred that processes that are not qualified as “substantial manipulation” are risk-free, notably if the processing of the cells entails long exposure of the cells/tissues to the environment. Accordingly, an analysis of the risks of the specific manufacturing process should be performed in order to identify the measures that are necessary to ensure the quality of the product. 简译: 1、不包括细胞/组织“实质性操作”的ATMPs生产工艺风险一般要低于涉及复杂“实质性操作”的ATMPs生产。但不能认为未认定为“实质性操作”的工艺没有风险,尤其要注意细胞的工艺是否有细胞/组织长时间暴露于环境的情况。 2、相应的,为了识别确保产品质量的必要措施,应当进行特定生产工艺的风险分析。 术语: substantial manipulation: 出自REGULATION (EC) No 1394/2007 Article 2(1)(c),从下文可以看出,所谓“substantial manipulation”是指为了实现与预期的再生、修复或替换相关的生物学特性、生理功能或结构特性进行的操作。例如基因操作(genetic manipulation),是对生物体的遗传物质进行人为的操作,使之发生修饰和改变的过程。 (c) Cells or tissues shall be considered ‘engineered’ if they fulfil at least one of the following conditions: - the cells or tissues have been subject to substantial manipulation, so that biological characteristics, physiological functions or structural properties relevant for the intended regeneration, repair or replacement are achieved. The manipulations listed in Annex I, in particular, shall not be considered as substantial manipulations, - the cells or tissues are not intended to be used for the same essential function or functions in the recipient as in the donor. REGULATION (EC) No 1394/2007 Annex 1 Manipulations referred to in the first indent of Article 2(1)(c)(不被视为“substantial manipulation”的操作) - cutting 切割 - grinding 研磨 - shaping 成型 - centrifugation 离心 - soaking in antibiotic or antimicrobial solutions 浸泡在抗生素或抗菌溶液中 - sterilization 灭菌 - irradiation 辐照 - cell separation, concentration or purification 细胞分离,浓缩或纯化 - filtering 过滤 - lyophilization 冻干 - freezing 冷冻 - cryopreservation 冻存 - vitrification. 玻璃化 总结观点: 1、“substantial manipulation”这个词直译过来是所谓“实质性操作”,从法规的描述来看,是通过一系列操作对细胞、组织等进行了实质性的改变。从REGULATION (EC) No 1394/2007 Annex 1这个列表上看,大多数物理加工或操作不属于实质性的改变。 2、无论其是否包括这种所谓“实质性操作”,均应当采取适当的风险降低措施来确保产品质量和用药安全。 2.45. With a view to reduce administrative burden, in the application of the GMP requirements to ATMPs the manufacturing process of which does not involve substantial manipulation, account may be taken of equivalent standards that are applied by ATMP manufacturers in compliance with other legislative frameworks. For instance, the premises and equipment that have been duly validated to process cells/tissues for transplantation purposes in accordance with standards that can be deemed comparable to those laid down in these Guidelines need not being validated again (for the same type of manufacturing operation). 简译: 从降低管理负担的角度来说,生产工艺中不包括“实质性操作”的ATMPs应用GMP要求时,ATMP生产企业可以考虑按照其他法规框架采用等同的标准。例如,出于移植目的,按照视同本指南的标准,使用已得到适当验证的设施设备来处理细胞/组织,不必再次验证(对同类型的生产操作)。 总结观点: 1、如果生产工艺中不包括“实质性操作”,换句话说就是低风险的ATMPs,也可采用其他与本GMP相同/等同标准的法规来指导生产。 2、这段的含义比较模糊,没有做出具体的要求和描述。不过从所举的例子来看,可能有下列的情况可参考: a、无菌保证:本GMP对无菌保证的要求与欧盟GMP Annex 1(征求意见稿)中的很多内容是相同的,因此执行欧盟GMP Annex 1(征求意见稿)中的要求也可符合本GMP; b、设备设施验证/确认:在同类型的生产操作中,例如在洁净区内使用同样的设备进行离心,如果设备设施已经进行了验证/确认且符合产品工艺的需求并在验证/确认的有效期内,那么这些已经完成验证/确认的设备设施可以不重新进行验证/确认。 2.46. However, there are certain elements of GMP that are intended to ensure the quality, safety and efficacy of the ATMPs which are not specifically addressed under other legislative frameworks and which, therefore, should follow the requirements in these Guidelines, also when the manufacturing process does not involve substantial manipulation. In particular, the requirements on product characterisation (through the setting of adequate specifications), process validation (the expectations for investigational ATMPs are described in Section 10.3), quality controls (in accordance with the terms of the marketing/clinical trial authorisation), and QP certification should be complied with. 简译: 1、但某些意图确保ATMPs质量、安全性和疗效的GMP要素在其他法规框架中没有明确规定,因此应当遵循本指南的要求,生产工艺中未包括“实质性操作”时也要遵循。 2、特别是应当符合对产品特征(通过设定恰当的质量标准),工艺验证(对临床用ATMPs的期望参见10.3),质量控制(按照上市许可/临床试验许可的条款)以及QP签发的要求。 总结观点: 1、紧接上一条,其含义就是如果某些操作或管理没有法规进行规定以及没有与本GMP相同或等同的标准/法规参考则应当执行本GMP,要满足本GMP的要求。 2、关于细胞治疗、基因治疗有很多法规,但不是每个法规都会对相关的GMP要素,例如降低混淆、差错、污染与交叉污染、不可控变化风险所采取的措施,会因硬件条件、产品特点的不同而不同,法规不可能照顾到每个细节,这时就需要参考本GMP的要求,适用时采取RBA进行管控。 2.47. ATMPs manufactured and applied during the same surgical procedure are not exempted from the ATMP Regulation (including therefore GMP compliance). 简译: 在同一手术过程中生产并使用的ATMPs不受ATMP法规的豁免(因此也包括GMP符合性)。 注:“the same surgical procedure”:细胞治疗的一种情况,即将某组织从病患身上取出并在同一手术过程中重新注入患者体内。 总结观点: lGMP的要求并不因产品的生产近于医疗手段而得到免除。 2.3.4. Additional considerations relevant for investigational ATMPs 2.48. While additional adaptations in the application of GMP may be justified in the case of investigational ATMPs, it is stressed that the quality, safety and traceability of the product should be ensured also in a clinical trial setting. 简译: 尽管临床ATMPs在实施GMP方面进行补充修正也许是合理的,但要强调的是产品的质量、安全性、疗效也应在临床试验环境得到保证。 总结观点: 尽管临床阶段GMP(I期)要求会“松”一些,但并不意味着可以牺牲产品的质量、安全性与疗效要求。换句话说,临床试验用样品要在符合GMP条件下进行生产和控制。 2.49. The following are examples of additional possible adaptations that may be acceptable in the case of investigational ATMPs: 简译:以下为也许可以接受的补充修正示例 2.50. • While investigational ATMPs should be manufactured in a facility with air quality requirements in accordance with the requirements set out in Sections 4.3.2 and 9.5, in case of investigational ATMPs in very early phase/proof of concept trials, it may be exceptionally possible to manufacture the product in an open system in a critical clean area of grade A with a background clean area of grade C if the following (cumulative) conditions are met: (i). A risk-assessment has been performed and demonstrated that the implemented control measures are adequate to ensure manufacture of the product of appropriate quality. In addition, the control strategy should be described in the investigational medicinal product dossier. (ii). The product is intended to treat a life-threatening condition where no therapeutic alternatives exist. (iii). The relevant competent authorities agree (agreement of both the assessors of the clinical trial and the inspectors of the site) 简译: 尽管临床用ATMPs应当在符合Sections 4.3.2 and 9.5中空气质量要求的厂房内生产,但如果符合下列条件,临床用ATMPs处于很早期/概念设计论证阶段时也许可在背景为C级,关键区域为A级的开放系统中生产。 (i). 已完成风险评估并证明了所执行的措施足以保证适当质量的产品的生产。另外,控制策略已在临床用药的文档中进行了描述。 (ii). 产品用于治疗没有取代品的危及生命的疾病。 (iii). 官方同意(临床试验评估员和现场检查员均应同意) 总结观点: 1、原则上,临床用ATMPs的无菌操作应当在B+A的环境或D级背景下使用隔离器进行。(Sections 4.3.2 and 9.5) 2、从后半句的字面上看,临床用ATMPs的生产环境放宽了要求,但是实际实施需要同时满足上述的三个条件,尤其是要获得官方的许可。企业并不能因为这段话,直接就将生产环境设计为C+A。 2.51. • In early phases of clinical research (clinical trial phases I and I/II) when the manufacturing activity is very low, calibration, maintenance activities, inspection or checking of facilities and equipment should be performed at appropriate intervals, which may be based on a risk-analysis. The suitability for use of all equipment should be verified before it is used. 简译: 1、在生产活动很少的临床研究(临床1期和1/2期)的早期阶段,厂房与设备应基于风险分析在适当的间隔内校准、维护、检查或核对。 2、所有设备的适用性应在使用前进行确认。 总结观点: 1、这里没有使用Qualification和Validation,意味着在I期临床以及临床I期/II期联合设计试验时,生产用厂房、设备需进行校准、维护与检查,而不必严格的进行验证/确认,但需要在使用前对设备的适用性进行测试,取得证明性资料,这个说法基本等同于进行设备的性能确认(PQ)。 2、上述做法的前提仍然是进行风险分析,不能没有任何依据就直接采用。 2.52. • The level of formality and detail for the documentation can be adapted to the stage of development. The traceability requirements should however be implemented in full. 简译: 文件的正式程度和详细程度应能够适于开发阶段。但应全部满足可追溯性的要求。 总结观点: 1、开发越向商业化生产靠近,体系的要求和文件的要求越来越严格,GMP的要素也越来越需要严格遵守。 2、无论是研发阶段、临床试验阶段还是商业化生产阶段,可追溯性的要求是一样的。扩大范围来说,数据完整性/可靠性的要求是一样的。 2.53. • During early phases of clinical development (clinical trial phases I and I/II) specifications can be based on wider acceptance criteria taking due account of the current knowledge of the risks and as approved by the competent authority that authorises the clinical trial. 简译: 在临床开发的早期阶段(临床1期和1/2期),质量标准可以建立在更宽泛的可接受标准的基础上,适当考虑现有的风险知识并经由准许临床试验的官方机构批准。 总结观点: 这段话可以用下面这张图来理解。
2.54. •Possible adaptations regarding qualification of premises and equipment, cleaning validation, process validation, and validation of analytical methods are described in Section 10. 简译: 关于对设施设备确认、清洁验证、工艺验证以及分析方法验证可能的修正参见Section 10.
|
| 【关闭】 |

